
概要
・中年期以降に発症し、淡蒼球、視床下核、小脳歯状核、赤核、黒質、脳幹被蓋の神経細胞が脱落し、異常リン酸化タウ蛋白が神経細胞内及びグリア細胞内に蓄積する疾患である(タウオパチーの1つ)
・病理学的には、アストロサイト内のtuft of abnormal fibers(tufted astrocytes)が、PSPに特異的な所見とされている。
・神経学的には、易転倒性、核上性注視麻痺、パーキンソニズム、認知症などを特徴とする。
・40歳以降、平均60歳代で発症する。男性に多く発症する。
・発症の原因は不明である。
症状
・初発症状はパーキンソン病に似ているが、安静時振戦はまれで、歩行時の易転倒性、すくみ足、姿勢保持障害が目立つ。
・進行するにつれて、頸部の後屈と反り返った姿勢、垂直性核上性眼球運動障害(初期には、眼球運動の随意的上下方向運動が遅くなり、ついには下方視ができなくなる。)
・構音障害
・嚥下障害
・想起障害と思考の緩慢を特徴とする認知症や注意力低下
・徐々に歩行不能、立位保持不能となって、寝たきりになる。
※最大の特徴は、初期からよく転ぶことである。著明な姿勢の不安定さに加え、注意力や危険に対する認知力が低下するため、何度注意を促してもその場になると転倒を繰り返す。
※バランスを失った時に上肢で防御するという反応が起きないため、顔面直撃による外傷を負うことが多い。
※注視麻痺は本症の特徴であるが、発症初期には認められないことが多い。下方視の障害が特徴で、発症3年程度で出現し、その後水平方向も障害される。
※筋強剛は四肢よりも頚部や体幹に強い。初期には頚部、四肢ともに全く筋強剛を認めず、むしろ筋トーヌスが低下していることがある。
※初期には姿勢がよく、頚部から下はまっすぐである場合が多い。一見無動にみえる患者が突然立ち上がったり、突発的な行動を起こすことがあるので注意が必要である。
※進行すると頚部が後屈する。
※認知症を合併するが程度は軽く、見当識障害や記銘力障害はあっても軽い。本疾患の認知症の本質は前頭葉の障害によるもので、把握反射、視覚性探索反応、模倣行動、使用行動などの前頭葉徴候が初期から出現する。動作の開始障害(無動、無言)、終了の障害(保続)などもよくみられる。
※様々な言語障害を合併する。嚥下障害は中期以降に出現することが多いが、早期に嚥下障害ある場合は生命予後が不良である。
検査所見
・頭部CTやMRIで脳幹,特に中脳あるいは橋被蓋部(橋の橋の背側)の萎縮,前頭葉の萎縮などがみられる。
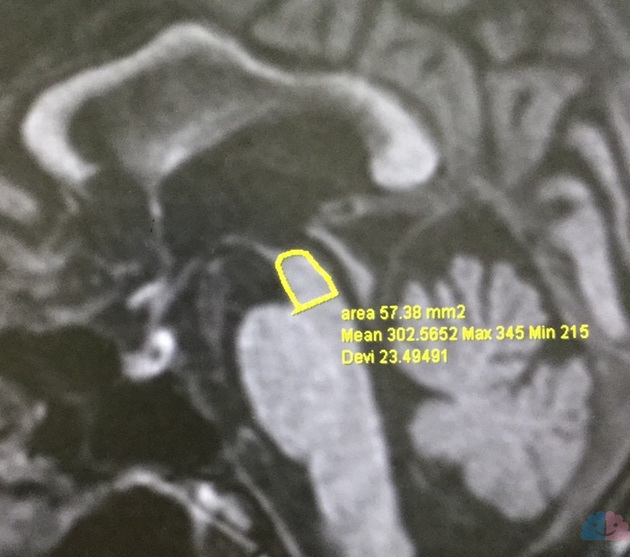
・決め手となるのはMRIの正中矢状断像における中脳被蓋部の萎縮で,「hummingbird sign」あるいは「penguin sign」と呼ばれている。
参照(このサイトより引用):https://www.ninchi-shou.com/entry/PSP-CTMRI-findings
MRI矢状断での中脳被蓋の面積低下
治療
・初期にはL-dopaが効く場合があるが、効果は長続きしない場合が多い。
・少量の抗コリン薬は無動に有効な場合が多いが、量が多いと突発的な行動が増えるので注意が必要である。
・抗うつ薬である塩酸アミトリプチリン、コハク酸タンドスピロンが奏功する場合もある。
・頚部・体幹のストレッチ運動、バランス訓練、言語訓練、嚥下訓練などのリハビリテーションを併用する。
予後
・ADL低下の進行は速く、我が国の剖検例の検討では車椅子が必要となるのに2~3年、臥床状態になるのに4~5年であった。
・平均罹病期間は5~9年という報告が多い。
・参考事項にあるパーキンソン病型や純粋無動症型は経過が緩徐で、罹病期間が10年以上であることも少なくない。
・死因は肺炎、喀痰による窒息などが多い。
コメント